Sickle Cell Anemia: Causes, Symptoms, and Treatment
21 Oct, 2023
 Healthtrip Team
Healthtrip TeamSickle Cell Anemia is a hereditary blood disorder characterized by abnormal hemoglobin, the protein responsible for carrying oxygen in red blood cells. This condition, named for the distinctive sickle shape of affected red blood cells, can lead to a range of health complications. Understanding the intricacies of Sickle Cell Anemia is crucial for both medical professionals and those affected by the condition.
Most popular procedures in India

Sickle Cell Anemia
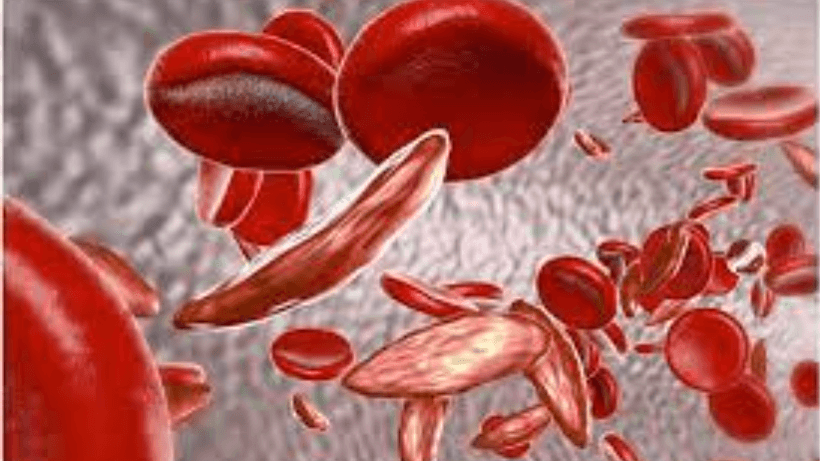
Sickle Cell Anemia is a genetic disorder that results from a specific mutation in the beta-globin gene of hemoglobin. Hemoglobin is a critical component of red blood cells, facilitating the transport of oxygen throughout the body. In individuals with Sickle Cell Anemia, the altered hemoglobin, known as hemoglobin S, causes red blood cells to take on a characteristic sickle shape. This abnormal shape hinders the cells' flexibility, leading to blockages in blood vessels, reduced oxygen delivery to tissues, and a range of associated health issues.
Wellness Treatments
Give yourself the time to relax
Lowest Prices Guaranteed!

Lowest Prices Guaranteed!
Demographics affected
a. Prevalence in Different Populations:
Sickle Cell Anemia exhibits significant variability in prevalence across different populations, primarily due to the historical distribution of the genetic trait and its association with regions where malaria is endemic.
- African Descent:
- Highest prevalence among individuals of African descent.
- Sub-Saharan Africa has the highest frequency of the sickle cell gene.
- Various subpopulations may show different prevalences and severity of the disease.
- Mediterranean and Middle East:
- Elevated prevalence in regions with a history of malaria, such as parts of the Middle East and the Mediterranean.
- India and Southeast Asia:
- Presence of the sickle cell gene in certain tribal populations.
- Geographic variations within the Indian subcontinent.
- Latin America:
- Prevalence in populations with African or Mediterranean ancestry.
- Distinct prevalence rates in different countries and ethnic groups.
b. Genetic Factors:
The prevalence of Sickle Cell Anemia is intricately tied to genetic factors, particularly the inheritance of the mutated hemoglobin genes.
- Autosomal Recessive Inheritance:
- Sickle Cell Anemia is inherited in an autosomal recessive manner, requiring the presence of two mutated hemoglobin genes (HbSS).
- Carriers (heterozygotes) with one normal and one mutated gene (HbAS) are referred to as having sickle cell trait and are generally asymptomatic.
- Consanguinity:
- Higher prevalence in populations where consanguineous marriages are common.
- Increased likelihood of both parents being carriers, leading to a higher probability of affected offspring.
- Malaria Resistance:
- The persistence of the sickle cell gene in populations is linked to its role in providing some resistance to malaria.
- Areas with historical malaria prevalence tend to have higher frequencies of the sickle cell gene.
Causes
- Genetic Mutation Explanation:
- Point Mutation in Beta-Globin Gene.
- Results in a change in the amino acid sequence.
- Production of abnormal hemoglobin known as hemoglobin S.
- Influence on Hemoglobin:
- Alters the structure of hemoglobin.
- Affects its ability to carry and release oxygen.
- Hemoglobin S tends to polymerize, leading to the characteristic sickle shape of red blood cells.
- Inheritance Patterns:
- Autosomal Recessive Inheritance.
- Requires two mutated hemoglobin genes (HbSS) for full expression.
- Carriers (HbAS) are asymptomatic.
- Consanguinity:
- Higher prevalence in populations with common consanguineous marriages.
- Increased likelihood of both parents being carriers.
- Malaria Resistance:
- The persistence of the sickle cell gene is linked to providing resistance against malaria.
- Higher frequencies in areas historically endemic to malaria.
Symptoms and Signs
a. Early Symptoms:
Sickle Cell Anemia manifests with a range of early symptoms, often appearing in childhood.
- Fatigue and Weakness:
- Due to the reduced oxygen-carrying capacity of the malformed red blood cells.
- Painful Episodes (Vaso-Occlusive Crises):
- Acute pain, often in the bones and joints, caused by the blockage of blood vessels by sickle cells.
- Paleness and Jaundice:
- Anemia leads to paleness, while the breakdown of red blood cells can cause jaundice.
b. Complications and Associated Signs:
The disease is associated with various complications that can have distinctive signs.
- Organ-Specific Complications:
- Spleen: Enlargement initially, but it may atrophy over time, increasing the risk of infections.
- Lungs: Acute chest syndrome, characterized by chest pain, cough, and difficulty breathing.
- Kidneys: Increased risk of kidney damage.
- Acute Complications:
- Pain Crisis: Intense, episodic pain requiring medical intervention.
- Stroke: Increased risk due to blood vessel blockage.
- Infections: Susceptibility to infections due to compromised immune function.
Understanding these symptoms and complications is crucial for timely diagnosis and management of Sickle Cell Anemia.
Diagnosis
The diagnosis of Sickle Cell Anemia involves a multifaceted approach, combining clinical assessments and specialized laboratory procedures.
a. Blood Tests and Laboratory Procedures:
- Hemoglobin Electrophoresis:
- This pivotal diagnostic tool discerns the types of hemoglobin present in the blood, highlighting the presence of abnormal hemoglobin S, the hallmark of Sickle Cell Anemia.
- Complete Blood Count (CBC):
- An essential test for evaluating anemia and providing insights into the quantity and characteristics of red blood cells.
- Reticulocyte Count:
- This test gauges the number of young red blood cells, serving as an indicator of the bone marrow's response to anemia.
- Peripheral Blood Smear:
- Microscopic examination of a blood sample reveals characteristic shapes and abnormalities in red blood cells, aiding in the identification of sickle-shaped cells.
- Bilirubin Levels:
- Elevated levels of bilirubin signify increased breakdown of red blood cells, a common occurrence in individuals with Sickle Cell Anemia.
- Blood Oxygen Levels:
- Assessment of blood oxygen levels, using methods such as pulse oximetry or arterial blood gas analysis, contributes to the diagnostic evaluation.
b. Genetic Testing:
- DNA Analysis:
- Definitive confirmation of Sickle Cell Anemia involves analyzing the patient's DNA to identify the specific genetic mutation responsible for the disorder.
- Prenatal Genetic Testing:
- Techniques such as chorionic villus sampling (CVS) and amniocentesis provide early detection of the mutation in developing fetuses, allowing for informed decisions.
- Newborn Screening:
- Routine screening programs at birth play a crucial role in the early detection of abnormal hemoglobin, enabling timely intervention.
- Confirmatory Tests:
- A comprehensive diagnosis often involves a combination of blood tests, genetic analysis, and clinical findings to ensure accuracy.
Treatment Options
a. Medications:
Several medications aim to manage symptoms and complications associated with Sickle Cell Anemia.
- Hydroxyurea:
- Stimulates the production of fetal hemoglobin, which is less prone to sickling.
- Reduces the frequency of pain episodes and acute complications.
- Pain Management:
- Analgesics for managing pain during vaso-occlusive crises.
- Nonsteroidal anti-inflammatory drugs (NSAIDs) for pain and inflammation.
- Antibiotics:
- Prophylactic use to prevent infections, given the increased susceptibility of individuals with Sickle Cell Anemia.
b. Blood Transfusions:
- Red Blood Cell Transfusions:
- Administered to increase the number of normal red blood cells.
- Used for managing severe anemia, preventing stroke, and treating complications like acute chest syndrome.
- Exchange Transfusions:
- Involves removing the patient's blood and replacing it with donor blood.
- Used to reduce the percentage of sickle cells and manage complications.
c. Bone Marrow Transplant:
- Allogeneic Transplant:
- Involves replacing the patient's bone marrow with that of a compatible donor.
- A potential cure for Sickle Cell Anemia, but the procedure carries significant risks and is not widely available.
- Gene Therapy:
- An emerging approach involves modifying the patient's own bone marrow cells outside the body to produce normal hemoglobin.
- Offers the potential for a more widely applicable and less risky treatment option.
Risk Factors
a. Genetic Factors:
- Homozygous Inheritance (HbSS):
- Individuals inheriting two mutated genes (HbSS) are at the highest risk of severe manifestations.
- Increased risk when both parents are carriers.
- Compound Heterozygotes (e.g., HbSC, HbS? Thalassemia):
- Depending on the specific variant, risks and severity may vary.
- Understanding the genetic background is crucial for predicting the course of the disease.
b. Geographic Considerations:
- Malaria-Endemic Regions:
- Presence of the sickle cell gene is associated with a reduced risk of severe malaria.
- Explains the higher prevalence of the gene in historically malaria-endemic areas.
- Access to Healthcare:
- Disparities in healthcare access impact the management and outcomes of Sickle Cell Anemia.
- Limited access may result in delayed diagnosis and suboptimal care.
- Consanguinity Rates:
- Populations with higher rates of consanguineous marriages may have an increased prevalence of the disease.
- Consanguinity contributes to a higher likelihood of both parents being carriers
Outlook/Prognosis
Life Expectancy:
Advancements in medical care have significantly improved the life expectancy of individuals with Sickle Cell Anemia. While life expectancy can vary based on factors such as the severity of the disease and access to healthcare, early diagnosis, comprehensive management, and preventive measures contribute to better outcomes. Regular medical follow-ups, including monitoring for complications, can further enhance life expectancy.
Quality of Life Considerations:
Quality of life for individuals with Sickle Cell Anemia is influenced by various factors:
- Pain Management:
- Effective pain management is crucial for improving daily functioning and overall well-being.
- Access to appropriate pain relief measures, including medications and supportive care, is essential.
- Education and Support:
- Education about the condition and its management empowers individuals to actively participate in their care.
- Emotional and psychological support, both from healthcare providers and a supportive community, plays a vital role in enhancing quality of life.
- Access to Comprehensive Care:
- Access to comprehensive healthcare, including specialized clinics and multidisciplinary teams, positively impacts the management of complications and overall health.
In conclusion, while challenges persist, advancements in understanding and managing Sickle Cell Anemia offer hope for better outcomes and an improved quality of life for individuals and families affected by this genetic blood disorder. Ongoing research is vital for further breakthroughs in treatment and care.
Related Blogs

Healthtrip: Your Guide to Leading Multi-Organ Transplant Centers
Healthtrip
Healthtrip: Advanced Brain Treatment Options with Expert Surgeons
Healthtrip

Healthtrip: Global IVF Treatment - Journey to Parenthood
Your Path to Parenthood with Healthtrip
Healthtrip: Navigating International Liver Transplant Options & Prices
Healthtrip
Healthtrip: Top 10 Countries for Liver Transplant Medical Tourism in 2025
Healthtrip Medical Tourism
Healthtrip: Top 15 Liver Transplant Surgeons for International Patients
Healthtrip





